SUPPLEMENTAL
DOCUMENT SD-4
Measurement
Uncertainty for Purity Determinations in Seized Drug Analysis
FOR PART IVC –
Quality Assurance/Uncertainty
Table of Contents
A Example 1: Control Chart Method. 2
A.1 Sampling Uncertainty Considerations. 3
A.2 Methodology. 3
A.3 Uncertainty Budget Table. 5
A.4 Calculation of combined standard
uncertainty. 6
A.5 Calculation of expanded uncertainty. 6
A.6 Results. 7
B Example 2: Use of Proficiency Test
Data to Estimate Uncertainty in Combination with Control Chart Data. 8
B.1 Sampling Uncertainty Considerations: 8
B.2 Methodology. 9
B.3 Uncertainty Budget Table. 10
B.4 Calculation of combined standard
uncertainty. 11
B.5 Calculation of expanded uncertainty. 11
B.6 Results. 12
C Example 3: Single Case Quantitation
Utilizing Replicate Samplings. 13
C.1 Sampling Uncertainty Considerations. 13
C.2 Methodology. 13
C.3 Conditions. 14
C.4 Calculation of combined standard
uncertainty. 15
C.5 Calculation of expanded uncertainty. 15
C.6 Results. 16
D
References. 17
D.1
Books. 17
D.2 Journal articles and reviews. 17
D.3 On-line resources. 18
Introduction
The
following examples demonstrate different approaches for deriving an estimate
for the uncertainty associated with purity determinations. In purity determination, uncertainty arises
from two factors: sampling (including sample homogeneity) and the analysis. Each factor is considered in the following
examples.
These
examples are meant to be illustrative, not exclusive. Laboratories should develop defensible
procedures that fit their operational environment and jurisdictional
requirements.
In
these examples, purity values are expressed as a percent composition by
weight. This can create confusion in calculations
where an uncertainty contribution is provided as a relative percent. Notes and example calculations are provided to clarify these applications.
Definitions
for the terms used can be found in the SWGDRUG
glossary Part IV C, Annex A or references listed below. The references also
contain additional examples and detailed information regarding estimation of
uncertainty.
The
following examples should not be directly applied to methodology used without
first considering the specific purpose of a method and its relevant operational
environment. It is assumed that
quantitative methods have been validated as per SWGDRUG
recommendations.
Scenario:
The laboratory is required to determine the percent purity of a single
bag of solid material, weighing approximately 3 grams and previously determined
to contain cocaine. In this example, measurement uncertainty is calculated
using control chart data obtained from a measurement assurance process that
mimics casework samples as closely as possible. Sample homogenization is tested to ensure the
laboratory’s homogeneity criteria are met.
After all of the
solid material has been homogenized, duplicate samples are collected and
analyzed. The laboratory has determined
that the purity difference between these two results must be equal to or less
than the control limits (± 3 standard deviations) from the control chart. This
process is used to establish the homogeneity of the sample.
The control chart
derived from the analysis of a measurement assurance sample is used to capture
contributions from analyst, matrix, environment, and other factors. The use of the measurement assurance sample
control chart will inevitably result in some contributions being counted twice
but these are difficult to isolate. This
is a conservative approach but the likely overestimation of uncertainty was
deemed acceptable by the laboratory.
The laboratory is
using a validated method that has been shown to have no significant bias. A method uncertainty was established as part
of the method validation and has a value of 0.9%relative. For this example, the laboratory established
this value by analyzing 5 different samples with known purity. These five samples were analyzed over five
days to establish figures of merit such as repeatability.
The laboratory uses a
solid commercial reference material for preparation of calibration solutions.
The analysis report states the purity to be ≥ 99.0% and the reference
material is stored as per manufacturer’s recommendation.
Long term variability
associated with method performance is captured by the control chart. The standard
deviation obtained from the control chart is calculated from the 100 most
recent points, which in this example is 2.1%relative. Including both
the control chart and method validation contributors will result in some
overlap, which is again accepted as a conservative approach.
Verification of
homogeneity:
Sample #1 purity:
27.8%purity
Sample #2 purity: 28.5%purity
Average: 28.2%purity
|27.8%purity
- 28.5%purity|/28.2% *100 = 2.5%relative
This value is smaller
than the control limits (±3SD or 3 x 2.1 = 6.3%relative) established
by the control chart, demonstrating acceptable homogeneity.
These results are
interpreted to mean that the variability seen between the two duplicate samples
is less that the variability of the validated method as demonstrated by the
control chart. The sample is then
acceptably homogeneous and the duplicate results can be averaged to obtain a
reported value.
If the difference
between these replicate values falls outside the range established by the
control chart, the bulk sample is considered to be inhomogeneous. In this instance, the bulk sample must be
further homogenized or the uncertainty budget would have to be adjusted to account
for the inhomogeneity.
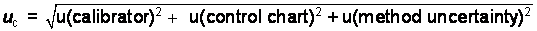
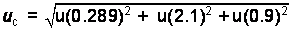

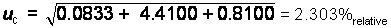
The
expanded uncertainty is expressed mathematically as:
U = k*uc
In this
case the uc is calculated
as a relative percentage and must be converted to an absolute percent of the
experimentally determined mean value:
2.303/100
= 0.02303 * experimentally determined value = 0.02303 * 28.2%purity
= 0.6494%purity
Using a
coverage factor k = 2 (confidence
level of approximately 95%, assuming the %purity follows a normal distribution[1]):
U = 2 * 0.6494%purity
= 1.30%purity
Using a
coverage factor k = 3 (confidence
level of approximately 99% assuming the %purity follows a normal
distribution):
U = 3 * 0.6494%purity
= 1.95%purity
A.6.1 28.2%purity
± 1.3%purity (k=2)
A.6.2 28.2%purity
± 2.0%purity (k=3)
Scenario:
The scenario is the same as in Example A, except this example also includes
contributions for uncertainty associated with reference material and matrix
effects using historical proficiency test data collected over several years
across a multi-laboratory system. This is a conservative approach but the
likely overestimation of uncertainty is deemed acceptable by the laboratory.
The
proficiency test is performed annually across multiple laboratories which use
different analytical techniques and different batches of reference materials
for quantitation. The controlled substance in the reference material is
cocaine. The proficiency data set represents 6 test events over the past six
years with 22 participating laboratories. In this example, the consensus value
is used as the accepted value for the %purity of the proficiency test sample.
Data
for this example is shown in the following table. All purities are given in units of %purity.
|
|
All participating laboratories
|
Laboratory #1
|
|
Year1
|
Consensus
value (Cref)2
|
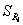 (%)3 (%)3
|
Proficiency test result
|
Bias (% error relative to the assigned
value)4
|
|
2012
|
17.9
|
4.8
|
18.7
|
+4.5
|
|
2011
|
29.3
|
2.6
|
28.8
|
-1.7
|
|
2010
|
23.3
|
7.9
|
24.2
|
+3.9
|
|
2009
|
26.1
|
5.1
|
26.0
|
-0.4
|
|
2008
|
13.7
|
9.5
|
13.3
|
-2.9
|
|
2007
|
33.5
|
3.3
|
34.1
|
+1.8
|
|
Mean:
|
5.55
|
|
|
1. For
each year, m = 22 participants
2.
Consensus value (mean) of %purity obtained from proficiency test
event
3. Reproducibility
standard deviation expressed as relative percentage values. This is the standard deviation for the %purity
values obtained within a given year for m = 22
4. Sample
calculation for 2012: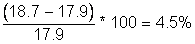
5. Arithmetic
mean reproducibility standard deviation (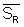 ) calculated over i years.
) calculated over i years.
The scenario is the
same as in Example A.
The scenario is the
same as in Example A. The calibrator
contribution is retained because Laboratory #1 is using a different reference
material for calibration for this case sample as was used for the proficiency
tests and for the control chart.
This example includes
two additional uncertainty contributions:
1) Method bias for
Laboratory #1: A contribution derived from Laboratory #1’s performance on the
proficiency tests as compared to the consensus values.
2) Standard
uncertainty of the mean () value: A contribution from the uncertainty associated with
the consensus values of the proficiency tests.
Information regarding
the determination of these two contributions and how they are evaluated may be
found in the references below
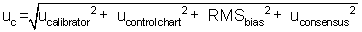
In
this example, the contribution from the calibrator could be excluded because of
its minimum contribution to the total combined uncertainty. The decision to remove a contribution should
be made after all uncertainty contributions are evaluated.
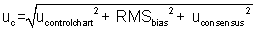
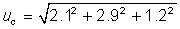
uc = 3.8%relative
Expanded
uncertainty is expressed mathematically as:
U
= k*uc
In
this case, uc is
calculated as a relative percentage and must be converted to an absolute percent
of the experimentally determined mean value:
3.8/100 = 0.038 * experimentally
determined value =
0.038
* 28.2%purity = 1.1%purity
Using
a coverage factor k = 2 (confidence
level of approximately 95% and assuming the %purity follows a normal distribution[3]):
U
=2*1.1%purity = 2.2%purity
Using
a coverage factor k = 3 (confidence
level of approximately 99% assuming the %purity follows a normal distribution):
U = 3*1.1%purity
= 3.3%purity
B.6.1 28.2%purity ± 2.2%purity (k=2)
B.6.2 28.2%purity ± 3.3%purity (k=3)
Scenario: The laboratory is required to determine the
percent purity of a single item of solid material, weighing 3 grams and
previously determined to contain heroin. The laboratory has a documented sampling plan
and although it does not routinely quantitate heroin, the laboratory has a method
available for general drug quantitation using a multipoint calibration curve. Homogeneity of the bulk case sample is not
assumed and variations in composition will be reflected in the standard
deviation of the replicates that is included in the combined uncertainty.
The
laboratory elects not to use a control chart.
Two samples are taken from a QC material with a known 79.3%purity. These two samples are prepared such that the
concentration of one is near the lower limit of the working range and the concentration
of the other is near the upper limit of the working range. The target analyte concentration in the case
samples must fall within the range bracketed by these two QC samples. All samples (calibration, QC and case) are
analyzed contemporaneously. The difference in the experimentally determined
purities of both QC solutions must meet pre-defined laboratory criteria.
In
this example the laboratory has elected to analyze 6 samples of the case
material.
The
laboratory has a general acceptance criterion for accuracy of ± 5.0%relative
for purposes such as method validation.
In this example, the laboratory has set the acceptance criteria for the
QC solutions to be ± 5.0%relative difference between the known value
and the experimentally determined value.
Six samples are taken from case
materials to account for variations in the composition of the bulk.
Uncertainty is estimated by
considering contributions from two sources:
1) The accuracy of the quantitative procedure as verified by the results
for the two QC solutions; and 2) the variability of the six samples.
It is assumed that if the
measured purity for the QC solutions both fall within ± 5.0%relative
of the accepted true value, the maximum contribution to uncertainty arising
from the method accuracy is ± 5.0%relative. In this example, acceptable accuracy was
demonstrated as follows:
Calibration curve
range: 0.100 mg/ml – 1.500 mg/ml
QC sample accepted
true value (%purity): 79.3
5%relative
of 79.3% = 4.0%
Acceptable range of QC
Sample results: 75.3 - 83.3%
|
|
Amount
Weighed / Volume
|
Experimentally
Determined Concentration
|
Calculated
Percent Purity of QC Sample
|
|
QC Solution
1
|
26.0
mg / 100 ml
|
0.214
mg/ml
|
82.3%
|
|
QC Solution
2
|
186.7
mg / 100 ml
|
1.423
mg/ml
|
76.2%
|
Experimentally
determined %purity Solution 1: 82.3% ACCEPTED
Experimentally
determined %purity Solution 2: 76.2% ACCEPTED
Working
range determined by QC solutions: 0.214 mg/ml – 1.423 mg/ml
Because both calculated
%purity values fall within ± 5.0%relative, a conservative approach
is taken to set method accuracy at 5.0%.
Had either or both of these
values fallen outside of the ± 5.0%relative range, the laboratory would
have to reassess the approach and procedure.
Results for sample
replicates:
|
|
% purity
|
|
Sample 1
|
26.0
|
|
Sample 2
|
24.9
|
|
Sample 3
|
25.0
|
|
Sample 4
|
27.0
|
|
Sample 5
|
25.4
|
|
Sample 6
|
27.0
|
|
Mean
|
25.88
|
|
s
|
0.947
|
|
Relative standard deviation
|
3.66%
|
In this example, the
laboratory elects to consider the tolerance value of ±5.0%relative as a
rectangular distribution in which a =
5.0 and
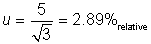
This is taken to represent
the contribution from the method bias.
Including this contribution is a conservative approach which will likely
result in an overestimation of the uncertainty.
It should be noted
that other ways exist of calculating and expressing the systematic component of
error for this analysis.
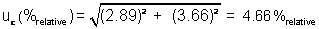
In this case, the expanded
uncertainty is calculated using the Student’s t value rather than k because the number of samples analyzed is
relatively small.
In this example, the
contribution from the systematic component, 2.89, is treated as a constant. As a result, the value of the Student’s t is determined based only on the
degrees of freedom associated with the random component determined by analysis
of the 6 replicates, or at 5 degrees of freedom.
This represents a
conservative approach. The Student’s t
value is selected at the 95% and 99% confidence levels.
In
this case, uc is calculated
as a relative percentage and must be converted to an absolute percent of the
experimentally determined mean value:
4.7/100
= 0.047 * experimentally determined value =
0.047
* 25.9%purity = 1.2%purity
95% confidence level
U = t*uc
U = 2.571 * 1.2%purity
= 3.1%purity
99% confidence level:
U = 4.032 * 1.2%relative
= 4.8%relative
Alternatively,
a laboratory may elect to calculate the effective degrees of freedom
to determine the value of the Student’s t,
which will fall between 2 and 2.571.
Selection of the value of 2.571 based on 5 degrees of freedom in this
case will likely result in an overestimation of the uncertainty.
C.6.1 25.9% ± 3.1% (95% confidence level)
C.6.2 25.9% ± 4.8% (99% confidence level)
D.1.1
Dieck, R.H. 2007. Measurement Uncertainty: Methods and Applications. 4th
ed. Research Triangle Park,
NC: The Instruments, Systems, and
Automation Society (ISA).
D.1.2 Kirkup, L., and R. B. Frenkel. 2006. An Introduction to Uncertainty in Measurement.
Cambridge, UK:
Cambridge University Press.
D.1.3 Taylor, J.R. 1997. An Introduction to Error Analysis: The Study of Uncertainties in
Physical Measurements. Sausalito,
CA: University Science Books.
D.2.1 ASTM, Standard Practice for Estimating
and Monitoring the Uncertainty of Test Results of a Test Method in a Single
Laboratory Using a Control Sample Program. ASTM International: West Conshocken, PA,
2007; Vol. E2554-07.
D.2.2 Baldan, A.; van der Veen, A. M. H.; Prauss,
D.; Recknagel, A.; Boley, N.; Evans, S.; Woods, D., Economy of proficiency
testing: reference versus consensus values. Accredit. Qual. Assur. 2001,
6 (4-5), 164-167.
D.2.3 Brown, R. J. C., and M. J. T. Milton. 2007.
Developments in accurate and traceable chemical measurements. Chemical Society
Reviews 36 (6):904-913.
D.2.4 Burns, M. 2004. Current practice in the
assessment and control of measurement uncertainty in bio-analytical chemistry.
Trac-Trends in Analytical Chemistry 23 (5):393-398.
D.2.5 Konieczka, P. 2007. The role of and the place
of method validation in the quality assurance and quality control (QA/QC)
system. Critical Reviews in Analytical Chemistry 37 (3):173-190.
D.2.6 Linsinger, T. P. J. 2008. Use of recovery and
bias information in analytical chemistry and estimation of its uncertainty
contribution. Trac-Trends in Analytical Chemistry 27 (10):916-923.
D.2.7 Meyer, V. R. 2007. Measurement uncertainty.
Journal of Chromatography A 1158 (1-2):15-24.
D.2.8 Populaire, S., and E. C. Gimenez. 2006. A
simplified approach to the estimation of analytical measurement uncertainty.
Accreditation and Quality Assurance 10 (9):485-493.
D.2.9 Pozivil, M., W. Winiger, S. Wunderli, and V.
R. Meyer. 2006. The influence of climate conditions on weighing results.
Microchimica Acta 154 (1-2):55-63.
D.2.10 Rios, A., and M. Valcarcel. 1998. A view of
uncertainty at the bench analytical level. Accreditation and Quality Assurance
3 (1):14-19.
D.2.11 Wong, S. K., Purity verification and
measurement uncertainty. Accredit. Qual. Assur. 2010, 15 (6), 337-341.
D.3.1 European Co-Operation for
Accreditation, Publication EA 4/02, Expression of the Uncertainty of
Measurement in Calibration, Dec 1999.
http://www.european-accreditation.org/n1/doc/ea-4-02.pdf
D.3.2 EURACHEM. Quantifying Uncertainty in
Analytical Measurement, 2007. http://www.measurementuncertainty.org/index.html.
D.3.3 JCGM. JCGM 100-2008: Evaluation of
measurement data (GUM 1995 with minor corrections). JCGM: Joint Committee for
Guides in Metrology, 2008. http://www.iso.org/sites/JCGM/GUM/JCGM100/C045315e-html/C045315e.html?csnumber=50461.
D.3.4 NIST 2007 Technical Note 1297, Guide for
Evaluating and Expressing the Uncertainty of NIST Measurement Results National
Institute of Science and Technology,
U.S. Department
of Commerce.
http://www.nist.gov/pml/pubs/tn1297/index.cfm
D.3.5 NIST. 2006. NIST/SEMATECH e-Handbook of
Statistical Methods: National Institute of Science and Technology, U.S.
Department of Commerce. http://www.itl.nist.gov/div898/handbook.
D.3.6 NIST. 2006. The NIST Reference on Constants,
Units, and Uncertainty 2009. Available from
http://physics.nist.gov/cuu/Uncertainty/index.html.
D.3.7 NIST 1994. NIST Technical note 1297Guidelines
for Evaluating and Expressing the Uncertainty of NIST Measurement Results.
National Institute of Science and Technology,
U.S. Department
of Commerce. http://physics.nist.gov/Pubs/guidelines/contents.html.
End of Document
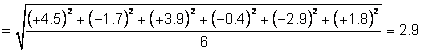
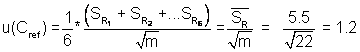 ;
;